














Huntington's Disease: Understanding the Genetic Neurodegenerative Disorder
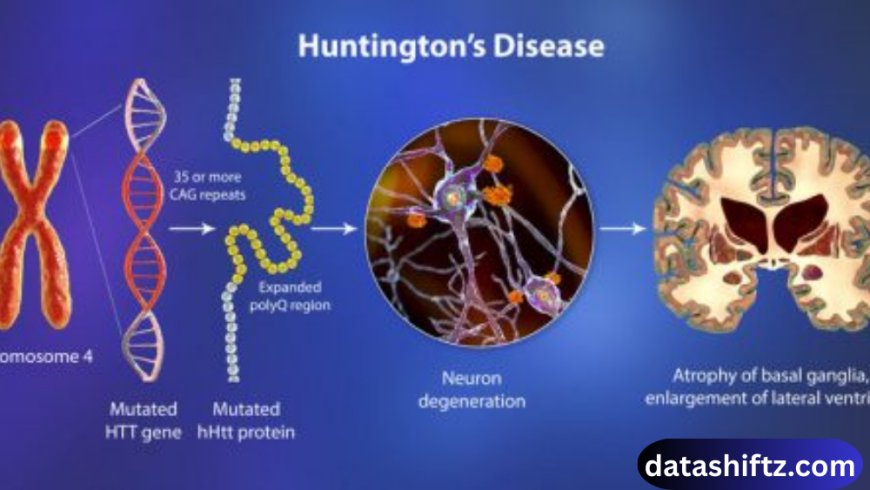
Introduction
Huntington’s disease (HD) is a rare, inherited neurodegenerative disorder that causes the progressive breakdown of nerve cells in the brain. Characterized by motor dysfunction, cognitive decline, and psychiatric symptoms, it affects thousands of people worldwide. First described by Dr. George Huntington in 1872, the disease remains incurable, but research continues to improve understanding, diagnosis, and management.
This article explores the causes, symptoms, diagnosis, treatment options, and ongoing research related to Huntington’s disease, offering a comprehensive guide for patients, families, and caregivers.
Causes and Genetic Basis
Genetic Mutation
Huntington’s disease is caused by a mutation in the HTT gene on chromosome 4, which encodes the huntingtin protein. The mutation involves an expansion of CAG trinucleotide repeats:
-
Normal individuals: 10–35 repeats.
-
Huntington’s patients: 36 or more repeats.
The longer the repeat, the earlier the onset and severity of the disease, a phenomenon known as anticipation.
Inheritance Pattern
HD follows an autosomal dominant pattern, meaning a child has a 50% chance of inheriting the disease if one parent carries the mutated gene. Both males and females are equally affected.
Symptoms of Huntington’s Disease
Huntington’s disease progresses gradually, often presenting in mid-adulthood (30s to 50s), although juvenile cases can occur. Symptoms are typically classified into three categories:
Motor Symptoms
-
Chorea: Involuntary, jerky movements affecting limbs, face, and trunk.
-
Dystonia: Muscle rigidity and abnormal postures.
-
Coordination Problems: Difficulty walking, speaking, and swallowing.
Cognitive Symptoms
-
Memory loss and difficulty planning or organizing.
-
Impaired judgment and problem-solving skills.
-
Slowed processing of information, impacting daily activities.
Psychiatric Symptoms
-
Depression, anxiety, and irritability.
-
Mood swings and impulsive behavior.
-
Obsessive-compulsive tendencies or social withdrawal.
Early recognition of these symptoms is essential for timely intervention and supportive care.
Stages of Huntington’s Disease
| Stage | Duration (approx.) | Key Symptoms | Functional Impact |
|---|---|---|---|
| Early Stage | 2–5 years | Minor chorea, subtle cognitive changes | Can maintain work and daily routines |
| Middle Stage | 5–10 years | Increased motor dysfunction, cognitive decline | Difficulty with daily activities, need for support |
| Late Stage | 10–15 years | Severe chorea, rigidity, cognitive impairment | Fully dependent on caregivers |
Diagnosis
Clinical Evaluation
Diagnosis begins with a neurological examination assessing motor skills, reflexes, and coordination.
Genetic Testing
-
DNA testing confirms the presence of the HTT mutation.
-
Genetic counseling is recommended for individuals with a family history of HD.
Imaging and Lab Tests
-
MRI or CT scans may reveal brain atrophy, particularly in the caudate nucleus and putamen.
-
Lab tests rule out other conditions with similar symptoms.
Early diagnosis enables better planning, symptom management, and participation in clinical trials.
Treatment and Management
Current Treatments
While there is no cure for Huntington’s disease, treatments focus on symptom management:
-
Medications for Chorea: Tetrabenazine and deutetrabenazine reduce involuntary movements.
-
Psychiatric Medications: Antidepressants, antipsychotics, and mood stabilizers manage psychiatric symptoms.
-
Physical Therapy: Improves mobility, balance, and muscle strength.
-
Speech and Occupational Therapy: Helps maintain communication and daily activities.
Lifestyle and Supportive Care
-
Regular exercise and nutrition help maintain physical health.
-
Support groups provide emotional and social support for patients and caregivers.
-
Advanced care planning addresses long-term needs and quality of life.
Emerging Therapies
-
Gene Silencing and Editing: Techniques like antisense oligonucleotides (ASOs) aim to reduce mutant huntingtin protein levels.
-
Stem Cell Therapy: Research explores replacing damaged neurons to restore function.
-
Clinical Trials: Patients may access experimental treatments targeting disease progression.
Key Facts About Huntington’s Disease
-
Autosomal dominant disorder, meaning only one copy of the mutated gene is sufficient to cause the disease.
-
Caused by a CAG repeat expansion in the HTT gene on chromosome 4.
-
Progressive neurodegeneration affects motor, cognitive, and psychiatric functions.
-
Symptoms often appear in mid-adulthood, but juvenile cases are possible.
-
Chorea is a hallmark symptom, alongside muscle rigidity and coordination issues.
-
Diagnosis is confirmed through genetic testing and supported by neurological exams.
-
There is no cure, but treatments manage symptoms and improve quality of life.
-
Supportive therapies include physical, occupational, and speech therapy.
-
Emerging therapies focus on gene silencing and neuroprotection.
-
Genetic counseling is essential for families with a history of HD.
Living with Huntington’s Disease
Coping with HD involves a multi-faceted approach:
-
Patient Education: Understanding the disease helps with planning and symptom management.
-
Family Support: Caregivers benefit from education, respite care, and counseling.
-
Community Resources: Organizations like the Huntington’s Disease Society of America (HDSA) offer advocacy, research updates, and local support networks.
-
Mental Health Care: Psychiatric support is crucial to address depression, anxiety, and behavioral changes.
A structured care plan can help maintain independence and quality of life for as long as possible.
Conclusion
Huntington’s disease is a devastating genetic disorder, affecting movement, cognition, and mental health. Despite the absence of a cure, advances in genetic research, symptomatic treatments, and supportive care have improved the outlook for patients and families.
Understanding HD, recognizing early symptoms, and accessing multidisciplinary care are essential for managing the disease. With ongoing research in gene therapy, neuroprotection, and clinical trials, there is hope for slowing disease progression and improving quality of life for those affected by Huntington’s disease.
Awareness, early diagnosis, and continued research remain key to addressing the challenges posed by this complex neurodegenerative condition.












